
Promotor-Analyse
(Untersuchung der Bindestellen von Ahr1 anhand von ChIP-Seq Daten aus Candida albicans)
(Stand: April 2021)
Hintergrund
Candida albicans ist ein human-pathogener Pilz, der bei Menschen mit schwachem Immunsystem schwere Infektionen hervorrufen kann. Er kann in verschiedenen Formen wachsen: als Hefe, Pseudohyphen und Hyphen [1]. Diese morphologische Flexibilität und insbesondere die Hyphenform sind für die Virulenz des Pilzes entscheidend [2, 3]. In den vergangenen Jahren wurden mehrere Virulenzfaktoren von C. albicans identifiziert. Sie stehen oft in engem Zusammenhang mit dem Hyphenwachstum des Pilzes. Dazu zählt auch der Transkriptionsfaktor Ahr1, von dem vermutet wird, dass er mehrere Virulenz-relevante Gene über eine Bindung in deren Promotorregion reguliert.
"Chromatin Immunopräzipitation Sequenzierung" (ChIP-Seq)
Zwei C. albicans Stämme mit einem hyperaktiven Ahr1 wurden mittels ChIP-Seq analysiert [4]. Mit diesem Verfahren können Protein-DNA-Interaktionen bestimmt werden. Das Protein wird mit der DNA gemischt, die DNA fragmentiert und alle nicht gebundenen DNA-Stücke entfernt. Anschließend löst man das Protein wieder von der DNA und analysiert diese mittels Hochdurchsatz-Sequenzierung. Resultat dieser Analyse waren 325 Sequenzabschnitte, die potentielle Bindestellen von Ahr1 im C. albicans Genom repräsentieren. Durch Nutzung deren chromosomaler Position wurden 532 Gene auf beiden DNA-Strängen identifiziert, in deren Promotorbereich die Sequenzen lagen, die also potentiell durch Ahr1 reguliert werden.
Promotor-Analyse
Eine Region von 500 Basenpaaren um das Maximum jedes Peaks eines Sequenzabschnitts (also den Punkt in der Sequenz, der in der Sequenzierung am häufigsten vertreten war und somit als die wahrscheinlichste Bindestelle angesehen werden kann) wurde als Eingabe für das Online-Tool Meme-ChIP (v. 5.1.0) [5] genutzt. So konnte ein hoch signifikantes Motiv gefunden werden, was mit einem bereits bekannten Bindemotiv von Ahr1 übereinstimmt. Zur weiteren Bestätigung dieses Motivs wurde die Software MochiView (v. 1.46) [6] genutzt. Der integrierte Motiv-Finder detektierte bei einer Suche nach möglichen Bindemotiven in den Peak-Regionen ebenfalls das bereits bekannte Ahr1-Motiv, welches in Abbildung 1 zu sehen ist.
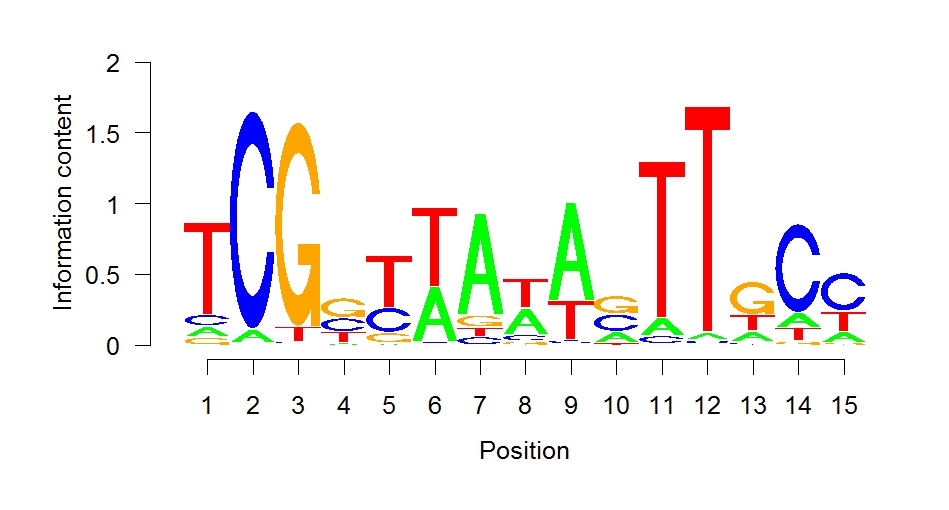
In MochiView können Peaks, Gene und Motive auch visualisiert werden. Es ist hierbei deutlich ersichtlich (Abbildung 2a und 2b), dass Ahr1 im Promotorbereich von wichtigen Virulenz-assoziierten Genen in C. albicans wie ECE1 und ALS3 bindet. Häufig ist mehr als eine potentielle Bindestelle pro Promotorregion vorhanden.
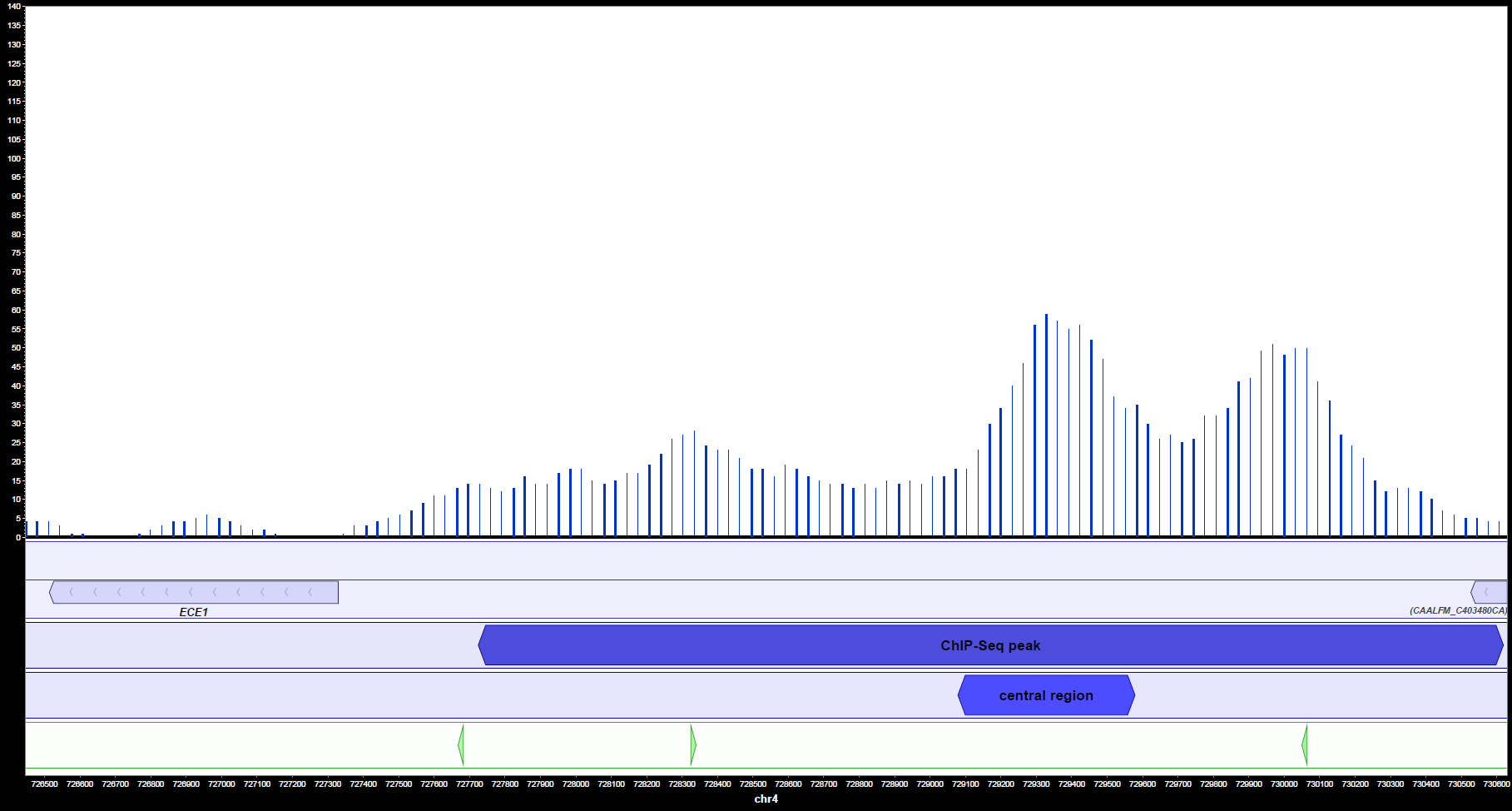
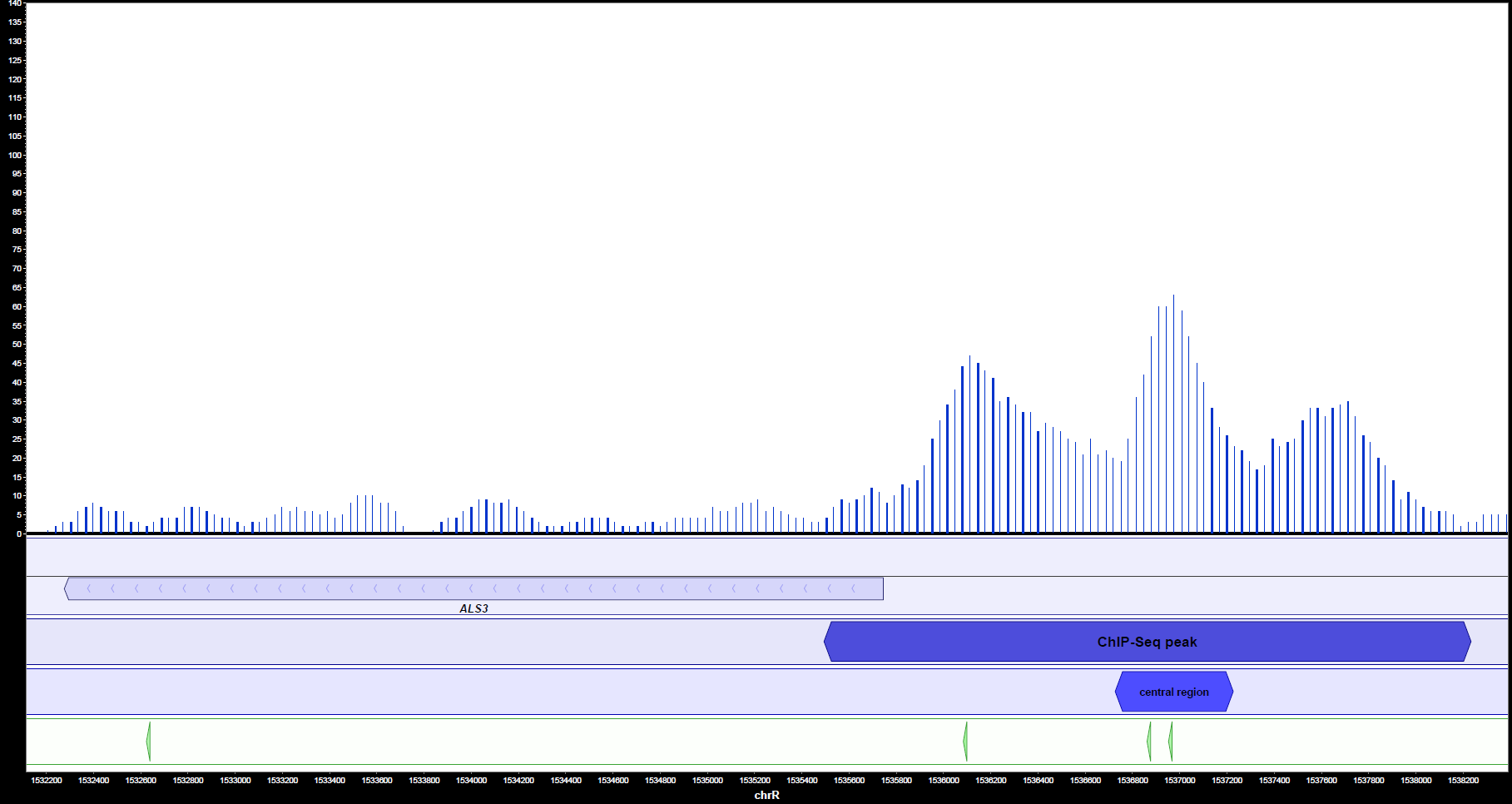
Insgesamt konnte das Bindemotiv in den Promotoren von 37 Virulenz-relevanten Genen nachgewiesen werden. Die unten stehende Tabelle zeigt für jedes dieser Gene jeweils das Motiv mit dem höchsten, von MochiView ermittelten Score. Falls es zwei Bindestellen mit gleichem Score gibt, sind beide aufgeführt.
| Gen-Name | Motiv-Entfernung zum Gen in bp | Motiv-Strang | Motiv-Score | Motiv-Sequenz |
|---|---|---|---|---|
| AHR1 | 5586 | - | 5.4 | GGCAACAATTACCGG |
| ALS1 | 1390 | - | 4.6 | GGAAACTTCAAACGA |
| ALS3 | 340 | + | 4.5 | TGCAAGTTAAACCGA |
| ALS4 | 1649 | - | 3.4 | CACAAGTGTAAGCGA |
| BCR1 | 2178 | - | 2.8 | AGAAAGGAAAAGCGA |
| BRG1 | 5765 | + | 5.1 | TGCAAGAATTACCGA |
| CDR1 | 1337 | + | 4.7 | ATCAACTATTGCCGA |
| CZF1 | 4304 | - | 2.4 | TGCAGTGGTAACCGA |
| DCK1 | 506 | + | 4.5 | GGAAAGTATAGTCGA |
| DEF1 | 1719 | - | 4.0 | GTCAACTTCTGACGA |
| DEF1 | 495 | - | 4.0 | GGAAATTAGAAACGA |
| EAP1 | 1204 | + | 4.5 | GGGAAGTTCAAGCGA |
| ECE1 | 2721 | + | 4.8 | GGGAAGAATTACCGA |
| EFG1 | 2311 | + | 5.0 | TGCAACTACAACCGA |
| FLO8 | 2066 | - | 3.5 | GGCAAGAAGTAGAGA |
| HGC1 | 11647 | - | 4.3 | GGAAAGTGGTAGCGA |
| HGT2 | 3442 | + | 4.9 | TGCAACTATTCGCGA |
| HWP1 | 1225 | - | 3.9 | GGCAAGTTTATCCGC |
| HYR1 | 1938 | + | 4.8 | AGCAATATTAGGCGA |
| IHD1 | 861 | + | 5.1 | TGCAACAATTACCGA |
| LMO1 | 179 | + | 4.4 | AGAAATTTTTAGCGA |
| MDR1 | 537 | - | 5.4 | GGTAACTATTGGCGA |
| NDT80 | 1567 | - | 5.1 | GGCAAGTTTAATCGA |
| RBT1 | 98 | - | 4.4 | GGTAAGATTTACCGG |
| RIM101 | 664 | - | 5.1 | AGCAAGTAGAGCCGA |
| SAP4 | 807 | - | 3.8 | AGCAATTTTAAGAGA |
| SAP5 | 1144 | + | 4.3 | GGCAATTTTAAGAGA |
| SAP6 | 833 | - | 3.5 | GGTAATTTTAAGAGA |
| SFL1 | 6551 | - | 5.1 | GGAAACTATTACCGG |
| SFL2 | 2733 | + | 4.0 | AACAAGTAGAGCCGA |
| SOD5 | 888 | + | 4.9 | GGCATCTTTTCCCGA |
| SOD5 | 1512 | + | 4.9 | GGAAAGTTGAAGCGA |
| STP2 | 915 | + | 2.9 | TGCAAGACTTGCAGA |
| TEC1 | 5005 | - | 4.8 | GGCAAGTATAAGCTA |
| UME6 | 15928 | + | 4.2 | AACAACTTTAAACGA |
| WOR1 | 5648 | - | 5.0 | AGCAAGTATAGCCGT |
| WOR2 | 1834 | + | 5.0 | GGCATCAATTACCGA |
Das weist darauf hin, dass Ahr1 an der Regulation einer Vielzahl von Virulenz-assoziierten Prozessen wie Hyphenbildung, Zellinvasion, Eisenaufnahme und Wirtszellbeschädigung beteiligt ist und durch die Bindestelle im eigenen Promotor möglicherweise sogar einen Feedback-Mechanismus aufweist.
Homologiesuche und Syntänie
Homologe von AHR1 (also Gene, die denselben Vorläufer haben), aber auch von ECE1 und ALS3 können in den Genomen von C. dubliniensis und C. tropicalis gefunden werden. Beide Arten sind eng mit C. albicans verwandt und ebenfalls pathogen. Die AHR1-Homologen beispielsweise haben in T-Coffee (v. 11.0) [7] einen sehr guten Alignment-Score von 992 (C. albicans AHR1 vs. C. dubliniensis Cd36_85930) bzw. 914 (C. albicans AHR1 vs. C. tropicalis CTRG_02263). ECE1 und seine Homologen kommen auf Scores von 994 (C. albicans ECE1 vs. C. dubliniensis Cd36_43260) bzw. 852 (C. albicans ECE1 vs. C. tropicalis CTRG_00476).
Auch die räumliche Lage der Gene im Genom (Syntänie) passt gut zueinander, wie man im Candida Gene Order Browser [8] sehen kann (in der Abbildung unten beispielhaft für AHR1), während sie zu S. cerevisiae, als nicht pathogenem Modellorganismus für Pilze, deutlich verschieden ist.
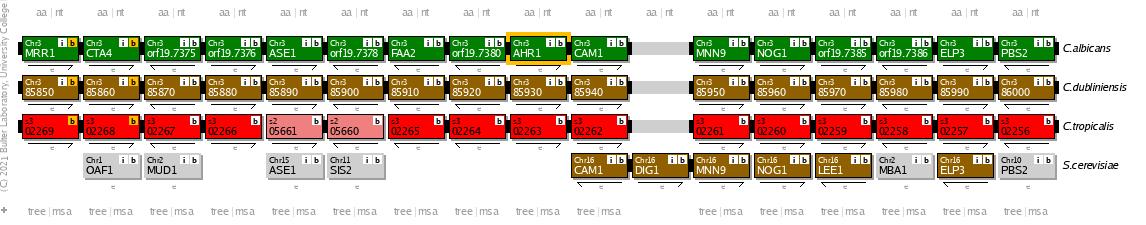
Möglicherweise gibt es also in diesen Spezies ähnliche Regulationsmechanismen wie in C. albicans.
Referenzen
- [1] P. E. Sudbery: Growth of Candida albicans hyphae. In: Nat. Rev. Microbiol., 9:737–748, 2011. doi: 10.1038/nrmicro2636
- [2] K. Zakikhany, J. R. Naglik, A. Schmidt-Westhausen, G. Holland, M. Schaller, B. Hube: In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. In: Cell Microbiol., 9:2938–2954, 2007. doi: 10.1111/j.1462-5822.2007.01009.x
- [3] A. L. Mavor, S. Thewes, B. Hube: Systemic fungal infections caused by Candida species: epidemiology, infection process and virulence attributes. In: Curr. Drug Targets, 6:863–874, 2005. doi: 10.2174/138945005774912735
- [4] S. Ruben, E. Garbe, S. Mogavero, D. Albrecht-Eckardt, D. Hellwig, A. Häder, T. Krüger, K. Gerth, I. D. Jacobsen, O. Elshafee, S. Brunke, K. Hünniger, O. Kniemeyer, A. A. Brakhage, J. Morschhäuser, B. Hube, S. Vylkova, O. Kurzai, R. Martin: Ahr1 and Tup1 contribute to the transcriptional control of virulence-associated genes in Candida albicans. In: mBio, 11:2, 2020.
doi: 10.1128/mBio.00206-20 - [5] P. Machanick, T. L. Bailey: MEME-ChIP: motif analysis of large DNAdatasets. In: Bioinformatics, 27:1696–1697, 2011. doi: 10.1093/bioinformatics/btr189
- [6] O. R. Homann, A. D. Johnson: MochiView: versatile software for genome browsing and DNA motif analysis. In: BMC Biol., 8:49, 2010. doi: 10.1186/1741-7007-8-49
- [7] C. Notredame, D. G. Higgins, J. Heringa: T-Coffee: A novel method for fast and accurate multiple sequence alignment. In: J. Mol. Biol., 302(1):205–217, 2000. doi: 10.1006/jmbi.2000.4042
- [8] S. L. Maguire, S. S. ÓhÉigeartaigh, K. P. Byrne, M. S. Schröder, P. O'Gaora, K. H. Wolfe, G. Butler: Comparative genome analysis and gene finding in Candida species using CGOB. In: Mol. Biol. Evol., 30(6):1281–1291, 2013. doi: 10.1093/molbev/mst042