
Vorverarbeitung von Omics-Daten
(Stand: November 2021)
In den vergangenen Jahren und Jahrzehnten ist eine große Bandbreite von Hochdurchsatz-(Omics-)Verfahren entwickelt worden, um biologische Systeme (z.B. Organismen, Krankheiten) besser zu verstehen. All diese Verfahren erzeugen eine große Menge an Rohdaten, die vor einer Analyse geeignet vorverarbeitet werden müssen. Dabei hängt es von vielen verschiedenen Faktoren ab, welche Vorverarbeitungsschritte das sind und wie sie durchgeführt werden müssen.
Experimentelle Planung
Vor der Durchführung aller Experimente muss das experimentelle Design gut durchdacht werden, denn es hat großen Einfluss auf die Vorverarbeitung. Es ist notwendig, genau zu bedenken, was die interessanten Messgrößen des Experiments sind (z.B. die Expression bestimmter Gene unter bestimmten Bedingungen) und was ggf. zusätzlich gemessen werden muss (z.B. die Expression aller Gene oder einiger sogenannter Housekeeping-Gene unter denselben Bedingungen). Man benötigt immer eine Messgröße, die im Experiment unverändert bleibt, um die Vorverarbeitung der Daten darauf auszurichten.
Auch die Anzahl der zu nehmenden Proben und deren Verteilung auf die verschiedenen experimentellen Bedingungen ist entscheidend für die Durchführbarkeit und die Art der Vorverarbeitung.
Die experimentelle Planung sollte also in jedem Fall gemeinsam von den ausführenden Wissenschaftlern und den Bioinformatikern/Datenanalysten durchgeführt werden um sicherzustellen, dass für die notwendigen Vorverarbeitungsschritte (und die statistische Auswertung) alle Bedingungen erfüllt werden.
Microarrays und 2D-Gele
Microarrays dienen vor allem der Bestimmung von Unterschieden in der Menge der mRNA aus zwei verschiedenen behandelten Zellen. Hierfür wird die mRNA in cDNA umgewandelt, die dann an immobilisierte DNA-Sonden auf den Microarrays bindet und über Fluoreszenz detektiert wird.
Ein Anwendungsbeispiel, in dem Vorverarbeitung und Analyse eines spezifischen Microarray-Datensatzes miteinander verbunden werden, befindet sich HIER.
2D-Gele kombinieren isoelektrische Fokussierung (IEF) mit orthogonal dazu ausgeführter SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) zur hochauflösenden Trennung komplexer Proteingemische in Einzelproteine. Sie werden beispielsweise genutzt, um Unterschiede im Proteingehalt zweier verschiedener Zellen zu untersuchen. Auch wenn heutzutage Massenspektrometrie-basierte Methoden einen großen Teil der Proteomics-Analysen ausmachen, hat 2D-Gelelektrophorese für einige Fragestellungen noch immer deutliche Vorteile [1].
Vorverarbeitung und Analyse eines 2D-Gel-Datensatzes wie im Folgenden beschrieben wurde von BioControl in der Vergangenheit [2] angewendet, wobei es sich bei diesem Beispiel um DIGE-Gele handelte, also Gele mit 3 Farbkanälen.
Bildanalyse
Kennzeichnende Eigenschaft für sowohl 2D-Gele als auch Microarrays ist, dass die ersten Vorverarbeitungsschritte bildbasiert sind. Hat man zwei oder mehr Proben, müssen die aufgenommenen Bilder der Microarrays oder Gele passgenau aufeinander gelegt werden um die jeweils gleichen mRNAs bzw. Proteine miteinander zu vergleichen. Bei Microarrays ist das recht einfach, da die Positionen der Sonden auf dem Array fest sind. Bei 2D Gelen bestimmen vielfältige Faktoren den genauen Verlauf der Proteintrennung, der jedes Mal ein wenig unterschiedlich ist [3].
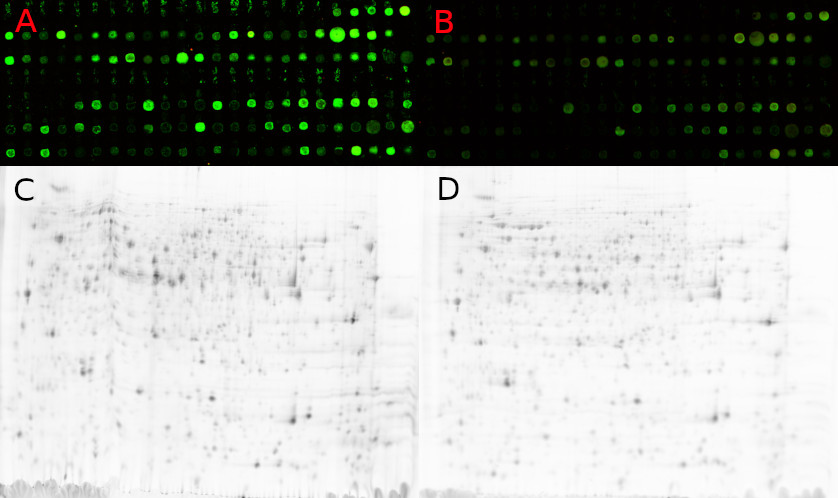
C,D – 2D-Gel-Scans zweier verschiedener Bedingungen, Spotpositionen variieren
(Bilder mit freundlicher Genehmigung des HKI Jena).
Das Matching der verschiedenen Bilder und die Spoterkennung übernimmt meist die Software, die mit dem Scanner zusammen geliefert wird. Wichtig sind hier die richtigen Einstellungen für Hintergrund-Normalisierung, Artefakt-Reduktion und, bei 2D-Gelen, die Trennung von Spotgruppen. Bei 2D-Gelen ist dabei in der Regel ein (iterativer) manueller Schritt notwendig, um das automatisierte Übereinanderlegen zu unterstützen und Fehler zu vermeiden. Da die kommerzielle Software recht teuer ist, gibt es auch immer wieder Versuche, mit Open-Source-Software ein ähnlich gutes Ergebnis zu erzielen [4].
Fehlende Werte
Probleme beim Matching von 2D-Gelbildern führen zu fehlenden Werten in den Daten der Protein Spots. Ein Protein ist dabei auf beiden Bildern vorhanden, wird aber (z.B. aufgrund von Verzerrungen) nicht richtig zugeordnet und so fehlen diesem Protein Werte bei bestimmten Bedingungen. Aber auch biologische Variabilität führt zu fehlenden Werten (sowohl bei Microarrays als auch bei 2D-Gelen), indem ein Gen bzw. Protein so reguliert ist, dass es in einer Probe auftaucht (also als Spot zu erkennen ist) und in einer anderen nicht.
Da fehlende Werte die spätere Analyse erschweren bzw. das Anwenden bestimmter Methoden (z.B. Clustering oder PCA) verhindern, muss man damit geeignet umgehen. Wenn fehlende Werte überwiegend biologisch verursacht sind, also das Gen bzw. Protein bei einer Bedingung gar nicht (oder unterhalb des technisch detektierbaren Bereichs) exprimiert bzw. vorhanden ist, kann man die fehlenden Werte durch einen Minimalwert ersetzen. Das kann z.B. der niedrigste gemessene Wert des Experiments sein, der ggf. mit einer gewissen Streuung belegt wird um die experimentelle Varianz widerzuspiegeln. Ist hingegen ein technisches Problem Ursache von fehlenden Werten, hat sich bei 2D-Geldaten ein k-nearest-neighbour Ansatz zur Imputierung bewährt [2].
Normalisierung
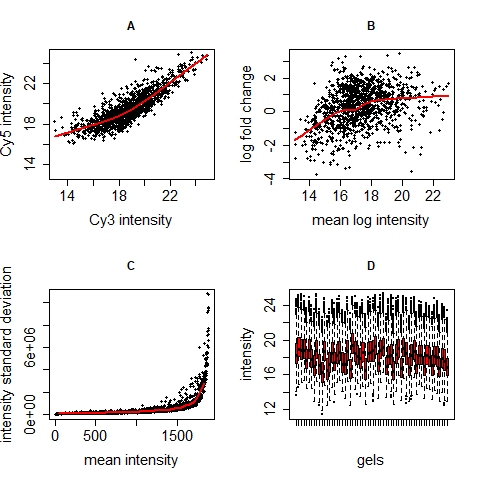
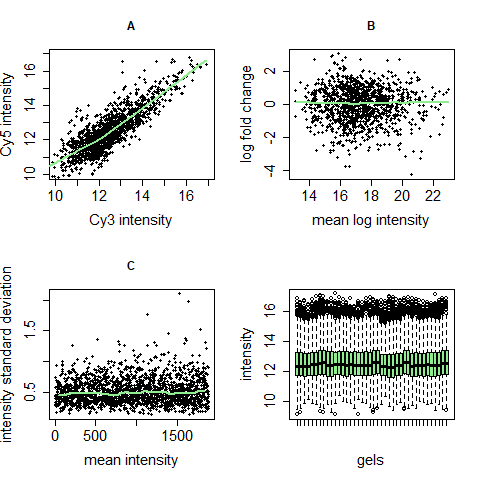
Für die Normalisierung von Microarray- und 2D-Gel-Daten gibt es sehr viele Möglichkeiten. Da beide Techniken auf Spots beruhen und ähnliche technische Probleme auftauchen, können viele Normalisierungsalgorithmen für beide angewendet werden. Wir zeigen hier eine Möglichkeit auf, die sich an vier spezifischen Problemen von Microarray- und 2D-Gel-Rohdaten orientiert und diese behebt [5]. Dafür werden zwei Normalisierungsschritte aufeinanderfolgend ausgeführt: Zentrierung und Varianzstabilisierung.
Grundlegende Hypothese für die Anwendung dieser Normalisierungstechniken ist, dass sich nur wenige Gene bzw. Proteine in ihrer Expression bzw. Regulation verändern, der Großteil aber gleich bleibt. Das ist in der Regel der Fall, wenn man das komplette Genom bzw. Proteom einer Zelle analysiert. Bei einer Analyse von nur einem Teil aller Gene bzw. Proteine kann diese Annahme verletzt sein und man benötigt eine andere stabile Messgröße, wie z.B. den Gesamtproteingehalt der Zelle.
Zentrierung und Varianzstabilisierung können vier in Microarray- und 2D-Geldaten häufig gesehene Probleme lösen:
- Farbstoffeffekte: Verschiedene Farbstoffe binden unterschiedlich an die Gene/Proteine und so kann ein Farbkanal im Scan heller sein als der andere, evtl. auch nur in bestimmten Intensitätsbereichen.
- Abhängigkeit der differentiellen Regulation vom Intensitätsmittel: Gene/Proteine mit geringerer Intensität sind nicht per se mehr oder weniger stark reguliert als Gene/Proteine mit höherer Intensität. Hier gibt es insbesondere im Bereich niedrigerer Intensitäten häufig Abweichungen.
- Abhängigkeit der Intensitätsvarianz vom Intensitätsmittel: Gene/Proteine mit höherer Intensität weisen häufig auch eine höhere Variabilität auf als solche mit niedriger Intensität obwohl sich das biologisch nicht erklären lässt.
- Unterschiedlichkeit der Intensitätsvarianz über verschiedene Gele: Unter der Annahme, dass die meisten Proteine eines Experiments nicht differentiell reguliert sind, sollten die Intensitäten in verschiedenen Gelen ähnlich verteilt sein, was in den Rohdaten oftmals nicht stimmt.
Next-Generation-Sequencing(NGS)- und Massenspektrometrie(MS)-Daten
NGS hat in vielen Anwendungsbereichen, wie zum Beispiel der Genexpressionsanalyse ganzer Genome, Microarrays weitgehend abgelöst, weil es hier schneller, kostengünstiger und genauer ist. Aus ähnlichen Gründen verwendet man heute in der Proteomik vielfach MS-basierte Methoden.
Fehlende Werte
NGS-Datensätze produzieren keine fehlenden Werte, wohl aber 0-Werte, wenn für ein Gen keine Fragmente gemessen bzw. zugeordnet worden sind. Für die Behandlung dieser Werte kann man je nach Anzahl verschieden vorgehen. Hat man bei 3 Replikaten nur 2 Werte, die größer als 0 sind, genügen diese meist für die folgenden Berechnungen. Bei einem vorhandenen Wert kann man den einen fehlenden Wert aus dem vorhandenen Wert und der lokalen Varianz im Datensatz berechnen. Bei 3 0-Werten imputiert man einen Wert z.B. mit dem niedrigsten gemessenen Wert unter Nutzung der lokalen Varianz.
Bei MS-Daten spielen 0-Werte oft eine große Rolle, insbesondere wenn nur Teile des Proteoms oder Metaboloms analysiert werden. Wenn es nur wenige fehlende Werte im Datensatz gibt, kann man sie mit geeigneten Minimalwerten imputieren und dabei ähnlich vorgehen, wie bei den NGS-Daten. Bei einer größeren Anzahl von 0-Werten, kann es notwendig sein, bestimmte Proteine oder ganze Proben aus der Analyse auszuschließen. Die ausgeschlossenen Proteine könnten dann mit einem Targeted-Ansatz nochmal gezielt untersucht werden indem ihre speziellen Bereiche im Spektrum genau analysiert werden.
Normalisierung
NGS-Daten müssen auf Sequenziertiefe und Genlänge normalisiert werden, um die unterschiedliche Anzahl an Reads pro Region im Genom zu kompensieren. Hierfür nutzt man beispielsweise RPKM-, TPM- oder MRN-Werte. Dadurch erreicht man eine Zentrierung der Daten, analog zur Normalisierung von Microarraydaten. Auf eine Varianzstabilisierung als zweiten Schritt kann man hingegen meist verzichten, die übernimmt das anzupassende (Fehler-)Modell. Notwendig ist sie in jedem Fall, denn bei NGS-Experimenten gibt es in den Bereichen mit niedrigen Intensitäten eine deutlich höhere Varianz als bei höheren Intensitäten.
Auch MS-Daten müssen normalisiert werden um Schwankungen in der technischen Ausführung der Analysen zu eliminieren. Hierbei ist es extrem wichtig, ob man ein gesamtes Zellproteom untersucht oder nur kleine Teile wie z.B. das Sekretom oder eine bestimmte Gruppe Metaboliten. So kann nicht immer das Gesamtproteom als Grundlage zur Normalisierung herangezogen werden. Möglicherweise eignet sich eher z.B. die Gesamtflüssigkeitsmenge einer Probe. Wenn auch das nicht gegeben ist, kann es dazu kommen, dass eine Normalisierung nicht möglich ist und die Rohdaten verwendet werden müssen. Auch das spricht dafür, dass sich die experimentell arbeitenden Wissenschaftler bereits vor dem Experiment gut mit den Bioinformatikern absprechen, denn diese Rohdaten müssen dann von besonders hoher Qualität sein um eine statistische Analyse zu ermöglichen.
Referenzen
- [1] K. Marcus, C. Lelong, T. Rabilloud: What room for two-dimensional gel-based proteomics in a shotgun proteomics world? In: Proteomes, 8(3):17, 2020. doi: 10.3390/proteomes8030017
- [2] D. Albrecht, R. Guthke, A. A. Brakhage, O. Kniemeyer: Integrative analysis of the heat shock response in Aspergillus fumigatus. In: BMC Genomics, 11:32, 2010. doi: 10.1186/1471-2164-11-32
- [3] D. Albrecht, O. Kniemeyer, A. A. Brakhage, R. Guthke: Missing values in gel-based proteomics. In: Proteomics, 10(6):1202–1211, 2010. doi: 10.1002/pmic.200800576
- [4] J. A. Molina-Mora, D. Chinchilla-Montero, C. Castro-Peña, F. García: Two-dimensional gel electrophoresis (2D-GE) image analysis based on CellProfiler: Pseudomonas aeruginosa AG1 as model. In: Medicine, 99(49):e23373, 2020. doi: 10.1097/MD.0000000000023373
- [5] D. Albrecht, O. Kniemeyer, A. A. Brakhage, R. Guthke: Normalisation of 2D DIGE data - On the way to a standard operating procedure. In: BIRD08, Schriftenreihe Informatik, 26:55–64, 2008.